随着大众健康意识的显著增强,食用植物油的营养价值、制作工艺、原料来源等品质受到越来越多的关注。受生产商广告影响,大众对广告中一再强调的“压榨”二字印象深刻,似乎压榨油才是好油,采用另一种主流工艺“浸出”生产的油就是品质有问题。但事实并非如此,目前国际上,包括发达国家在内,主要采用的生产工艺恰恰是“浸出”,即采用有机溶剂抽提粉碎后油料中的油脂,蒸馏回收有机溶剂后获得产品。浸出工艺有出油率高、生产成本低等优点,缺点是生产过程中不可避免的存在有机溶剂残留在产品中的情况,常用浸出溶剂中含有微量的苯、硫化物等有毒物质[1],溶剂残留量的控制好坏很大程度决定了浸出油的品质。2005年10月1日实施的国家标准GB2716-2005《食用植物油卫生标准》[2]中就严格规定了浸出油的溶剂残留量≤50 mg/kg,2018年12月21日实施的国家标准GB 2716-2018《食用植物油卫生标准》[3]中更是将溶剂残留量进一步降低为≤20 mg/kg。对应现行有效的国家标准检测方法为2017年6月23日实施的GB5009.262-2016《食品安全国家标准食品中溶剂残留量的测定》[4],方法相对2003版[5]最大的变化是定量采用了内标法。在对新方法进行确认过程中,注意到部分厂家生产的内标工作溶液溶剂N,N-二甲基乙酰胺引入的杂质干扰测定,且溶剂峰保留时间偏长,降低了样品检测效率。为避免干扰测定,同时缩短样本检测周期,提高样品检测效率,本研究查阅了大量国内外食用植物油中溶剂残留测定方法相关文献[6,7,8,9,10,11,12,13,14],最终参考美国油脂化学家协会(American oil chemists society,AOCS)溶剂残留测定方法[15],采用正庚烷纯品为内标,添加内标时改为使用1μL注射器加入0.5μL,并采用全自动顶空进样器进行样品前处理,对改进后方法的线性范围、检测限、精密度、回收率等指标进行了评估,发现各项指标均等同或优于原方法,适用于食用植物油中溶剂残留的测定,为相关检测提供参考。
2 材料与方法
2.1 仪器与试剂
气相色谱仪配氢火焰离子化检测器,;DB-5柱(30 m×0.25 mm,0.25μm,);7697A全自动顶空进样系统(美国安捷伦公司);E100H超声波清洗机(德国Elma公司);BT224S电子天平(德国Sartorius公司)。
“六号溶剂”(标准物质,10 mg/mL,国家粮食与物资储备局科学研究院,批号:BW3599);正庚烷(色谱纯,上海化学试剂一厂)。
食用油为市售以及大豆油。
2.2 实验方法
2.2.1 仪器条件
初始柱温50℃,保持3 min后以1℃/min的速率升温至52℃,再以30℃/min的速率升温至200℃,并保持3 min;载气(高纯氮)流速设定为1 m L/min;进样口温度设定为250℃,分流比设为100∶1;检测器温度设定为300℃,氢气流速设定为30 m L/min,空气流速设定为300 m L/min。全自动顶空进样器设定平衡温度为55℃,平衡时间为30 min,振荡速率设定为250 r/min,进样体积设定为0.5 mL。
2.2.2 基体植物油制备
采用市售的一级大豆油,超声脱气处理至溶剂残留量低于检测限。
2.2.3 标准曲线绘制
取6只20 mL顶空进样瓶,分别称取5.0 g(精确至0.01 g)基体植物油加入顶空进样瓶,每瓶加入0.5μL正庚烷后轻摇混匀,再分别加入0、5、10、25、50、100μL“六号溶剂”标准溶液,迅速密闭后轻摇混匀,即得浓度为0、10、20、50、100、200 mg/kg的标准系列;制备好的标准系列按照上述仪器条件上机测定,以保留时间定性,以标准溶液与内标物浓度比为横坐标,标准峰总面积与内标峰面积比为纵坐标绘制标准曲线。
2.2.4 样品测定
称取5.0 g(精确至0.01 g)基体植物油样品至顶空进样瓶,再加入0.5μL正庚烷,迅速密闭后轻摇混匀,按照上述仪器条件上机测定。
3 结果与分析
3.1 色谱条件优化
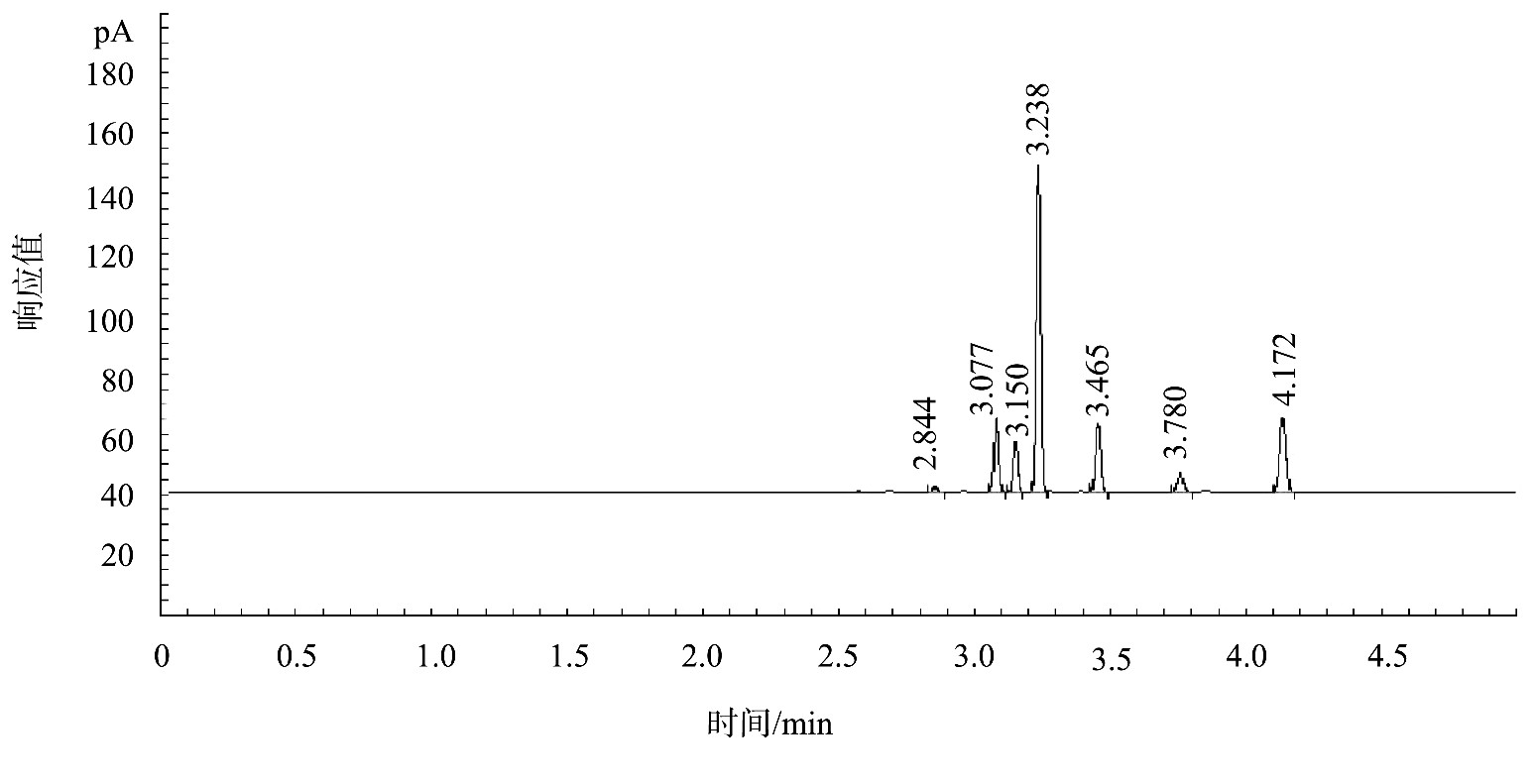
按照原方法提供色谱参考条件设定气相色谱仪,初始柱温设定为50℃,保持3 min后以1℃/min的速率升温至55℃,保持3 min后再以30℃/min的速率升温至200℃,并保持3 min,基体植物油标准色谱图显示保留时间最长的内标物正庚烷出峰时间为4.172 min。据此可将柱温条件优化为初始柱温50℃,保持3 min后以1℃/min的速率升温至52℃,再以30℃/min的速率升温至200℃,并保持3 min。柱温条件优化后,既保证了待测物和内标物色谱峰的分离,又将样品测定时间从19 min左右缩短到13 min左右,提高了样品检测效率。色谱图见图1。
3.2 内标添加方式优化
配制内标工作溶液时,为避免溶剂N,N-二甲基乙酰胺可能引入的杂质干扰测定,同时缩短样本检测周期,提高样品检测效率。通过查阅国内外文献、标准方法等资料,我们尝试参考美国油脂化学家协会(AOCS)溶剂残留测定方法,采用正庚烷纯品为内标,添加内标时改为使用1μL注射器加入0.5μL,优化后进一步考察方法性能指标。
3.3 标准曲线及检出限
按照上述条件进行标准曲线绘制,标准曲线线性方程为Y=1.93X+0.11,相关系数r=0.999,标准曲线见图2;按照GB/T 5009.1-2003《食品卫生检验方法理化部分总则附录A》[16]中对检出限的规定,计算优化后方法的检出限为0.5 mg/kg,定量限为1.6 mg/kg,详细数据见表1。
图1 基体植物油标准色谱图
图2 改进后方法标准曲线图
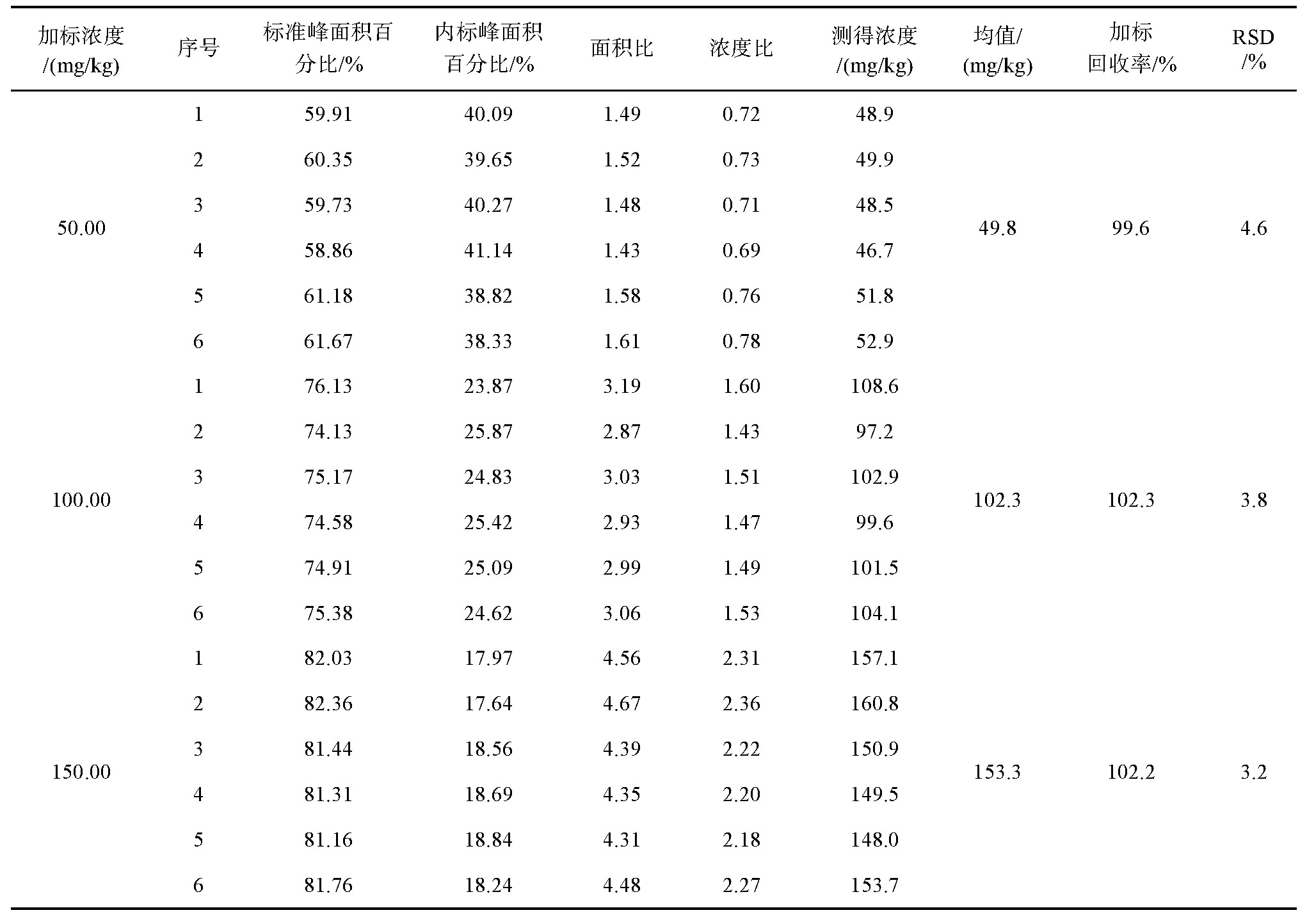
3.4 加标回收实验
按照GB/T 5009.1-2003中对精密度试验的要求,称取50 g基体植物油18份,分为3组,分别加入250、500、750μL“六号溶剂”标准溶液,混匀后即得浓度为50、100、150 mg/kg的标准样品,按照上述样品测定流程测定,测定结果算得相对标准偏差(relative standard deviation,RSD)为3.2%~4.6%,详细数据见表2。
4 结论
为避免溶剂杂志干扰测定,提高样品检测效率,本研究借鉴AOCS测定方法,改用正庚烷纯品为内标,并采用全自动顶空进样器进行样品前处理,同时根据出峰情况,优化了柱温条件,进一步提高了检测效率。改进后方法的线性范围为10~200 mg/kg,检出限为0.5 mg/kg,回收率为99.6%~102.3%,相对标准偏差为3.2%~4.6%,适用于大批量食用植物油中溶剂残留的测定。
表1 改进后方法检出限、定量限计算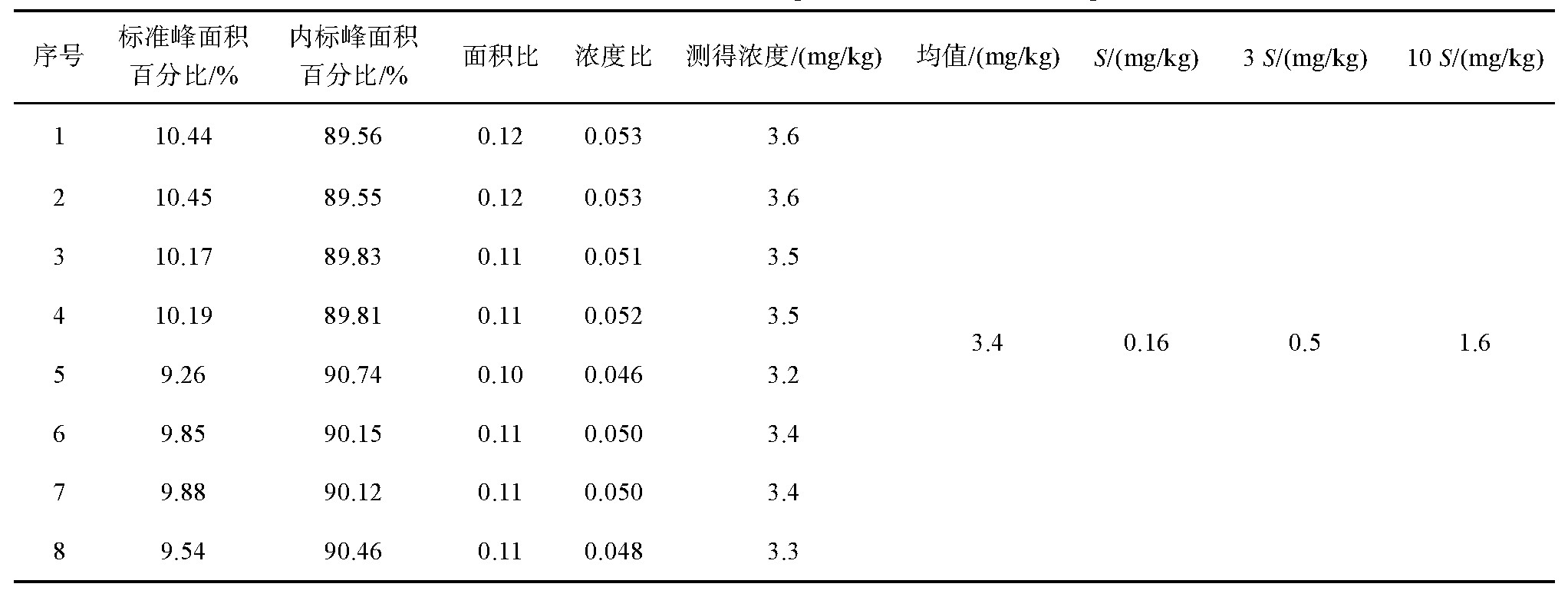
注:S为标准偏差,下同。
表2 改进后方法精密度试验(n=6)